
RNA sequencing is a technique that allows the analysis of gene activity in a sample, such as blood, brain, or other animal tissue. Currently, it is one of the most widely used tools in computational biology and medicine, as it facilitates the study of the impact of diseases on gene expression, which, in turn, affects protein synthesis and, consequently, cellular function.
Antes de 2009, se utilizaba la secuenciación en masa (Bulk RNA-seq), que proporcionaba un perfil global de expresión génica para toda la muestra combinada. El resultado de este método era un vector cuya dimensión correspondía al número total de genes de la especie, donde cada entrada representaba el nivel promedio de expresión de un gen en la muestra analizada. Sin embargo, en 2009 surgió una nueva tecnología llamada secuenciación de célula única (scRNA-seq), que se ha consolidado como una de las técnicas más utilizadas en el campo. Este método incluye un paso adicional para separar químicamente cada célula individual antes de la secuenciación, lo que permite analizar la expresión génica a nivel celular en lugar de hacerlo de manera global. Esto permite un análisis más detallado, especialmente en los distintos tipos celulares, que se caracterizan por sus propiedades fisiológicas, morfológicas y funcionales. Al obtener un perfil de expresión para cada célula, esta metodología genera una matriz en la que se presenta el nivel de expresión de cada célula individual (ver Figura 1).
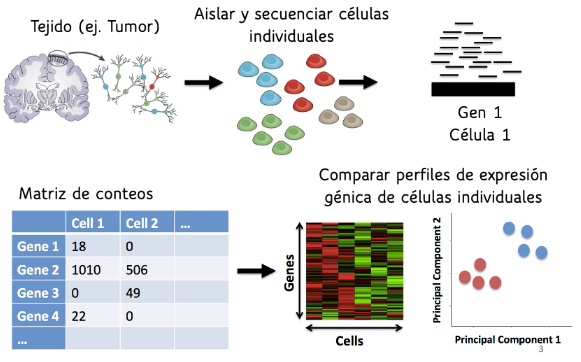
Figura 1: Pasos de Singel Cell RNA-seq
Una de las primeras tareas en el análisis de datos de scRNA-seq es la clusterización de las células, cuyo objetivo es agruparlas de manera que los grupos reflejen los tipos celulares presentes en la muestra. Este proceso enfrenta varios desafíos importantes:
Este proyecto se centró en la clusterización de los datos, proponiendo nuevas metodologías basadas en la distribución inherente de los mismos. Además, se realizaron comparaciones con métodos previamente utilizados y con enfoques tradicionales para la agrupación de datos en alta dimensionalidad. Finalmente, se desarrolló un demo de una aplicación que ejecuta los métodos más efectivos desarrollados en el proyecto.
La primera aproximación desarrollada utiliza un método de agrupación basado en grafos. A partir de la matriz de conteos, se calcula la correlación de Pearson entre cada par de células y se construye el grafo k-MST con k = log(número de células). Inicialmente, el grafo se inicializa como el árbol de expansión mínima (MST) del grafo original. Luego, se recalcula el MST del grafo inicial, excluyendo las aristas que ya han sido incorporadas en el nuevo grafo. Este proceso iterativo continúa durante k iteraciones, y el grafo final se obtiene al unir los k árboles de expansión mínima ortogonales. Una vez construido el grafo, se ejecuta el algoritmo de Louvain para detectar las comunidades (Figura 2).
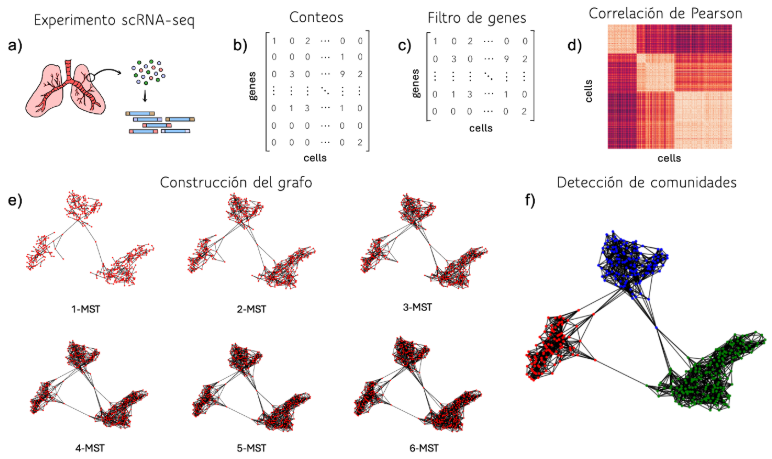
Figura 2: Algoritmo basado en grafos. a) Extracción de datos de células individuales. b) Consolidación de la matriz de recuentos crudos. c) Normalización de los datos basada en factores de tamaño. d) Cálculo de correlaciones de Pearson en pares. e) Construcción del grafo. f) Detección de comunidades.
La segunda aproximación utiliza una red de tipo autoencoder para la reducción de dimensionalidad y para aprender los parámetros de los clusters. A diferencia de una arquitectura tradicional, esta red no tiene solo una capa de salida, sino tres, que representan los parámetros de la distribución binomial negativa con inflación de ceros. A partir de esto, se define una función de pérdida llamada LZINB, que es de tipo log-likelihood y permite ajustar los datos a esta distribución. La binomial negativa se emplea comúnmente en datos de secuenciación debido a su capacidad para modelar la sobredispersión, mientras que la inflación de ceros facilita la modelación de los dropouts en los datos.
La segunda función de pérdida utilizada LGMM permite aprender los parámetros de los clusters mediante una función característica de los modelos de mezcla gaussiana, donde se asume que cada cluster sigue una distribución normal. El entrenamiento se divide en dos fases: la primera se enfoca en la reducción de dimensionalidad, mientras que en la segunda se introduce el aprendizaje de los parámetros de los clusters.
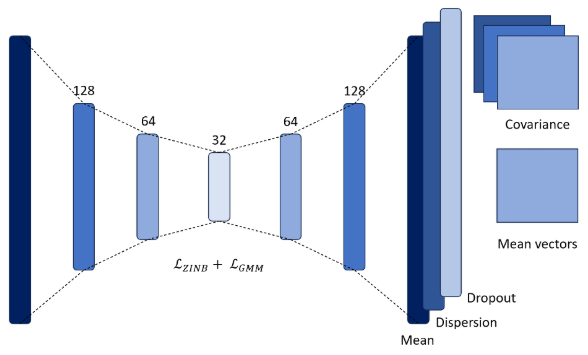
Figura 3: Arquitectura de la metodología basada en redes neuronales
Por último, se desarrolló un modelo supervisado utilizando la base de datos del Human Protein Atlas, que contiene perfiles de expresión para diferentes tipos celulares en 81 tejidos humanos. Aunque se podría utilizar cualquier modelo supervisado, más del 50% de los tipos celulares tenían una única representación, lo que complicaba la aplicación de métodos tradicionales. Por esta razón, se optó por un enfoque basado en Naive Bayes, utilizando una distribución binomial negativa para ajustar los conteos de la matriz de entrenamiento. Además, se incorporó información previa sobre el tejido secuenciado, dando más peso a los tipos celulares correspondientes al tejido analizado, lo que mejoró la precisión del modelo. Antes de entrenar el modelo, las bases de datos fueron normalizadas para asegurar que todas las características tuvieran la misma magnitud, facilitando así una comparación más equitativa entre las distintas variables.
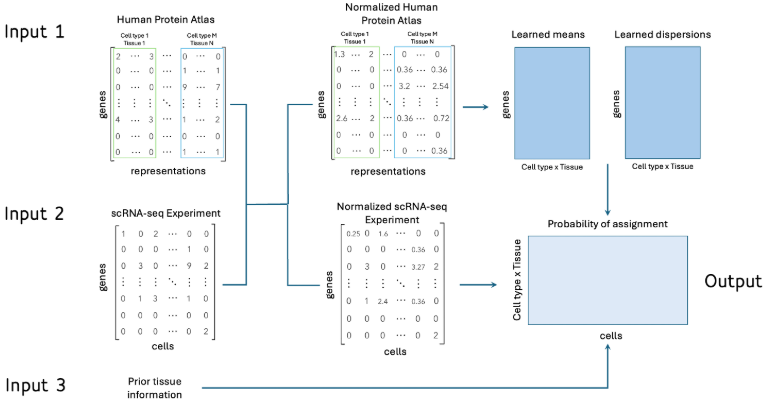
Figura 4: Algoritmo supervisado basado en la distribución binomial negativa. Se reciben tres entradas, la base de Human Protein Atlas (a), la matriz de conteos del experimento a analizar (b) y el tejido del experimento (c). Se realiza una normalización de los datos para garantizar que se mantengan las mismas magnitudes (d) y se procede a entrenar el modelo (e), en donde se aprenden las medias y dispersiones de cada gen en los tipos celulares considerados. Finalmente, con la base normalizada se obtienen las probabilidades de cada célula de pertenecer a los tipos celulares del HPA (f)
El rendimiento de los tres modelos se comparó con metodologías tradicionales para datos de alta dimensionalidad y con herramientas utilizadas actualmente para la clusterización de datos de scRNA-seq. Para esto, se utilizaron tanto datos simulados generados con la herramienta Symsim como datos de experimentos reales, comparando los clusters identificados por expertos con los detectados por los algoritmos. En la mayoría de los casos, al menos uno de los algoritmos desarrollados mostró mejores resultados que las otras aproximaciones. Sin embargo, ninguno de los métodos se destacó de manera consistente sobre los demás, posiblemente por la complejidad inherente de los datos. Esto resalta la necesidad de continuar investigando y desarrollando nuevas metodologías de agrupación para el análisis de datos de secuenciación.
Get information about Data Science, Artificial Intelligence, Machine Learning and more.
Link to the seminar presentation
https://www.youtube.com/watch?v=Q3MBYLpNDI0
References
In the Blog articles, you will find the latest news, publications, studies and articles of current interest.